Metabolism
on-line - the virtual tutorial room
copyright © 2008 - 2015 David A Bender
Two boys with profound fasting hypoglycaemia and no ketone bodies
LF is an 18 month old boy. He is the first child of healthy non-consanguineous parents, delivered after a normal pregnancy. He was well until he had an episode of gastroenteritis, leading to diarrhoea and vomiting for several days, when he was admitted to hospital in a coma, with a blood glucose concentration of 1.0 mmol /L. There was no smell of acetone on his breath, and on testing his urine with a dipstick there were no detectable ketone bodies. He regained consciousness after an intravenous infusion of glucose.
Later during his stay in hospital he was fasted under close supervision, with blood samples taken at intervals over 18 hours, by which time he had become hypoglycaemic, and was given an intravenous infusion of glucose. The following results were obtained on analysis of blood samples taken during his fast (all in mmol /L):
LF |
control subjects |
||||
13 h fasting |
15 h fasting |
17 h fasting |
18 h fasting |
24 h fasting |
|
| glucose | 4.1 |
3.8 |
2.9 |
2.3 |
3.6 ± 0.4 |
| non-esterified fatty acids | 2.38 |
2.48 |
3.34 |
3.96 |
1.2 ± 0.2 |
| beta-hydroxybutyrate | 0.03 |
0.05 |
0.05 |
0.02 |
2.0 ± 0.2 |
| acetoacetate | nd |
nd |
nd |
nd |
1.0 ± 0.2 |
nd = not detectable
(From data reported by Morris AAM, et al, Pediatr Res 44:3
392-6, 1998)
What conclusions can you draw from these results?
The first impression is that LF appears to have a similar problem to TFP, who was unable to metabolism long-chain fatty acids, and therefore became profoundly hypoglycaemic on fasting (see the exercise on "Muscle weakness and hypoketotic coma on fasting"). However, in this case we have no evidence of muscle weakness. While it is possible the LF cannot oxidise fatty acids, it is also possible that he is able to oxidise them, but cannot synthesise ketone bodies from the resultant acetyl CoA.
In order to determine whether his problem was in the oxidation of fatty acids, or the synthesis of ketone bodies from acetyl CoA, fibroblasts were cultured and incubated with [3H]fatty acids. The results show nmol [3H]water released per hour per mg protein for two replicate assays on LF's fibroblasts and mean ± sd (and range in parentheses) for fibroblasts from 20 control subjects.
LF |
control subjects |
||
| [9,10-3H]oleate | long-chain, C18:1 n-9 | 3.7, 4.0 |
4.5 ± 0.8 (3.0 - 6.1) |
| [9,10-3H]myristate | medium chain, C14:0 | 6.0, 6.3 |
5.4 ± 1.0 (3.8 - 7.1) |
| [9,10-3H]palmitate | long chain, C16:0 | 5.8, 6.3 |
4.7 ± 0.9 (3.1 - 6.7) |
(From data reported by Morris AAM, et al, Pediatr Res 44:3 392-6, 1998)
What conclusions can you draw from these results?
There is obviously no problem with LF's ability to oxidise fatty acids, so the problem must lie in the synthesis of ketone bodies from acetyl CoA.
The pathway of ketone body synthesis is shown below
 :
:
.
 Hydroxymethylglutaryl
CoA (HMG CoA) is an intermediate in the catabolism of the amino acid leucine.
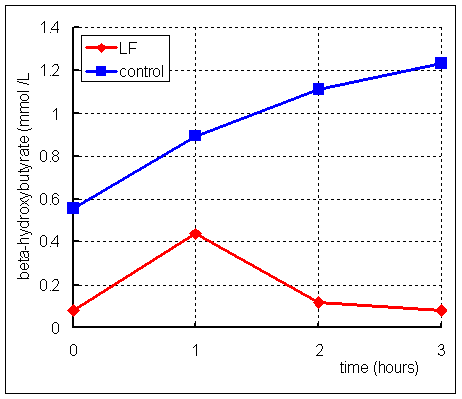
After an 11 hour fast, LF was given a dose of 200 mg /kg body weight leucine,
and blood samples were taken for measurement of beta-hydroxybutyrate. The results
are shown on the right:
Hydroxymethylglutaryl
CoA (HMG CoA) is an intermediate in the catabolism of the amino acid leucine.
After an 11 hour fast, LF was given a dose of 200 mg /kg body weight leucine,
and blood samples were taken for measurement of beta-hydroxybutyrate. The results
are shown on the right:
What conclusions can you draw from these results?
LF can obviously make beta-hydroxybutyrate from leucine, which means that he has adequate activity of HMG CoA lyase. Since he cannot make beta-hydroxybutyrate from fatty acids, this suggests that his problem is lack of either beta-ketothiolase or HMG CoA synthase. You do not know this at present, but beta-ketothiolase is also involved in the catabolism of ketone bodies, so it is most likely that he lacks HMG CoA synthase.
Why do you think that LF shows a rise in plasma beta-hydroxybutyrate after the leucine dose, followed by a fall, whereas in the control subject the plasma concentration of beta-hydroxybutyrate continues to rise throughout the 3 hours after the dose of leucine?
Both LF and the control subject are fasting. The control subject has a normal ability to synthesise ketone bodies from acetyl CoA, and as his fast continues so he synthesises more to provide a metabolic fuel for muscle, so as to spare glucose for the brain and red blood cells.
By contrast, LF cannot synthesise ketone bodies from acetyl CoA. However, the beta-hydroxybutyrate that he has synthesised from leucine is metabolised by muscle - this explains why his plasma concentration of beta-hydroxybutyrate falls again after the peak produced from leucine has been metabolised.
In the fasting state LF does have some plasma beta-hydroxybutyrate. What is the likely source of this beta-hydroxybutyrate?
In the fasting state there is more catabolism of tissue proteins than replacement synthesis. This is partly because of the high ATP cost of protein synthesis and partly because of the use of the amino acids released from tissue proteins being used for gluconeogenesis or as metabolic fuel. The most likely source of beta-hydroxybutyrate will be leucine liberated by the catabolism of tissue proteins. It is also possible that LF has some residual activity of HMG CoA synthase, so that he can synthesise a small amount of ketone bodies from acetyl CoA, but nowhere near enough to meet his energy needs in fasting.
What do you think is the advantage of reducing acetoacetate to beta-hydroxybutyrate in the liver?
You will see from the pathway of ketone body synthesis above that acetoacetate undergoes a non-enzymic decarboxylation to acetone. Acetone is poorly metabolised (if at all). Therefore formation of acetone represents a loss of (potentially valuable) metabolic fuel in the fasting state and starvation. Beta-hydroxybutyrate is stable, so reducing acetoacetate to beta-hydroxybutyrate minimises this loss.
Although acetone is metabolically more or less useless, clinically it is extremely useful, because it has a distinct smell, and can be detected on the breath. This provides a rapid way of telling that an unconscious person is ketotic, before blood or urine samples are tested for ketone bodies.
HL is a four and a half year old boy. When he was 6 months old he had an attack of vomiting and diarrhoea, became lethargic and dehydrated, and was taken to the emergency department in a coma. On admission his plasma glucose was 0.3 mmol /L, and he was hyperventilating; his plasma pH was 7.1 and plasma bicarbonate was 5 mmol /L. There was no smell of acetone on his breath, and on testing his urine with a dipstick there were no detectable ketone bodies. Analysis of a plasma sample for ketone bodies showed that none was detectable. He regained consciousness after an intravenous infusion of glucose, and bicarbonate to allow respiratory compensation for the metabolic the acidosis.
Analysis of a urine sample by hplc showed the presence of a number of abnormal organic acids, including methylglutaconic and hydroxymethylglutaric acids, and their carnitine conjugates. As shown in the pathway below, both of these are metabolites of leucine.
