Metabolism
on-line - the virtual tutorial room
copyright © 2008 - 2015 David A Bender
Muscle weakness, heart failure and profound hypoglycaemia in a young girl
CUD is a 7 year old girl. Since early infancy she has suffered from occasional attacks of coma, especially when she has been suffering from one of the usual childhood feverish illnesses. She tires easily, and has never been able to undertake strenuous exercise, or keep up with her peers in the playground.
Six months ago she complained of chest pains, and a physical examination was suggestive of heart failure, with tachycardia, moderate mitral insufficiency, gallop rhythm and left ventricular heave. A two dimensional echocardiogram showed the left ventricular end-diastolic dimension to be elevated at 7.46 cm, with a fractional shortening of 15%. A chest X-ray showed moderate enlargement of her heart. Her liver was moderately enlarged and palpable.
A fasting blood sample taken when she was first admitted to hospital gave the following results:
CUD |
reference range |
|
| glucose (mmol /L) | 2.6 |
3.8 - 6.0 |
| non-esterified fatty acids (mmol /L) | 1.8 |
1.0 - 1.4 |
| ketone bodies (mmol /L) | not detectable |
2.0 - 3.0 |
| sodium (mmol /L) | 141 |
135 - 145 |
| potassium (mmol /L) | 4.1 |
3.6 - 5.0 |
| bicarbonate (mmol /L) | 20 |
18 - 23 |
| ammonium (µmol /L) | 60 |
< 50 |
| pH | 7.4 |
7.35 - 7.45 |
| insulin (µU /mL) | 3.8 |
4 - 5 |
| glucagon (pg /mL) | 190 |
160 - 180 |
| aspartate aminotransferase (units /L) | 55 |
< 31 |
| alanine aminotransferase (units /L) | 64 |
< 31 |
| creatine kinase - muscle isoenzyme (units /L) | 366 |
< 170 |
| lactate dehydrogenase (units /L) | 250 |
< 290 |
| alkaline phosphatase (units /L) | 300 |
< 150 |
| gamma-glutamyl transpeptidase (units /L) | 290 |
< 60 |
| total carnitine (µmol /L) | 5.1 |
40 - 60 |
What conclusions can you draw from these results?
She is slightly hypoglycaemic, and her non-esterified fatty acids are slightly higher than would be expected for an overnight fast. The lack of detectable ketone bodies is interesting and will bear further investigation. Her plasma concentrations of insulin and glucagon are appropriate for her mild hypoglycaemia. Her plasma ammonium concentration is somewhat higher than normal.
The elevated activities of aspartate and alanine transaminases, alkaline phosphatase and gamma-glutamyl transpeptidase are all suggestive of liver disease or damage.
The elevated activity of creatine kinase is suggestive of muscle damage - either skeletal or cardiac muscle, but the high normal activity of lactate dehydrogenase suggests that there has not been any recent death of cardiac muscle cells.
The abnormally low serum concentration of carnitine is interesting.
 Carnitine
is both synthesised in the body and also obtained the diet. This means that
dietary carnitine deficiency is unlikely, and indeed strict vegetarians, who
have negligible dietary sources of carnitine are still able to maintain a normal
plasma concentration.
Carnitine
is both synthesised in the body and also obtained the diet. This means that
dietary carnitine deficiency is unlikely, and indeed strict vegetarians, who
have negligible dietary sources of carnitine are still able to maintain a normal
plasma concentration.
A urine sample showed that she excreted very much more carnitine than children of her age, and most of this was as free carnitine, not fatty acid esters of carnitine (acyl carnitine).
Her renal clearance of carnitine (both free and esterified) was almost the same as that of creatinine, whereas normally the renal clearance of free carnitine is only about 2% of that of creatinine (although esterified carnitine has a higher renal clearance than free carnitine).
What conclusions can you draw from these observations?
Carnitine is a small, water-soluble compound, and will therefore be more or less completely filtered at the glomerulus. The fact that the normal renal clearance of carnitine is only about 2% of that of creatinine suggests that there must be active reabsorption of carnitine in the renal tubules. If CUD's renal clearance of carnitine is more or less the same as her creatinine clearance then it seems likely that she has a defect of the carnitine transporter that is responsible for the reabsorption of carnitine in the renal tubules. Her very low plasma carnitine is therefore most likely the result of excessive loss in the urine rather than a defect in carnitine synthesis or a dietary deficiency.
While CUD was in hospital she developed a viral infection that led to a moderate fever. The next morning she was deeply unresponsive and could not be woken up. A blood sample taken at this time gave the following results:
| CUD | reference range |
|
| glucose (mmol /L) | 2.0 | 3.8 - 6.0 |
| non-esterified fatty acids (mmol /L) | 1.9 | 1.0 - 1.4 |
| ketone bodies (mmol /L) | not detectable | 2.0 - 3.0 |
| sodium (mmol /L) | 142 | 135 - 145 |
| potassium (mmol /L) | 4.2 | 3.6 - 5.0 |
| bicarbonate (mmol /L) | 20.5 | 18 - 23 |
| ammonium (µmol /L) | 150 | < 50 |
| insulin (µU /mL) | 3.6 | 4 - 5 |
| glucagon (pg /mL) | 191 | 160 - 180 |
What conclusions can you draw from these results?
Her coma is obviously due to both profound hypoglycaemia, and the significantly elevated ammonium concentration. (You will see in a later exercise how even a modest increase in plasma ammonium can lead to a disturbance of consciousness).
It is noteworthy that despite her profound hypoglycaemia, while plasma non-esterified fatty acids are higher than normal (suggesting that they are being mobilised from adipose tissue triacylglycerol), again she has no detectable ketone bodies. This suggests a defect in the synthesis of ketone bodies from fatty acids.
Why do you think her plasma ammonium is increased?
This is presumably the result of gluconeogenesis from amino acids, in an attempt to maintain an adequate plasma concentration of ammonium. Normally the ammonium released in amino acid catabolism is used for the synthesis of urea, which is excreted in the urine. Urea synthesis occurs in the liver, and we already have evidence from the enzyme data when she was first admitted to hospital that she has liver damage or disease.
What emergency treatment would you give her?
Since her main problem is profound hypoglycaemia, the obvious immediate treatment would be intravenous glucose. She responded well, and regained consciousness.
Why do you think she was so profoundly hypoglycaemic when she had a fever?
There is an increase in basal metabolic rate of about 13% for each degree C of fever - so a fever will increase her metabolic rate, and hence the rate at which she metabolises glucose - effectively the only fuel available to her tissues.
In order to investigate her muscle weakness and liver damage, biopsy samples were taken.
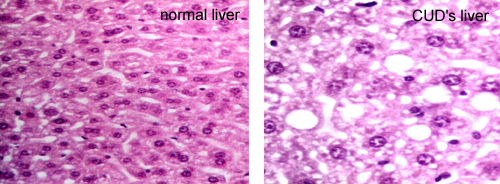 As
shown on the right, her liver showed evidence of fatty infiltration, with an
abnormal number of droplets of lipid in the liver, appearing in this slide as
empty vacuoles.
As
shown on the right, her liver showed evidence of fatty infiltration, with an
abnormal number of droplets of lipid in the liver, appearing in this slide as
empty vacuoles.
Her muscle biopsy also showed deposition of abnormal amounts of lipid between the fibres.
What conclusions can you draw from these results?
It seems likely that although she can mobilise non-esterified fatty acids from adipose tissue in the fasting state, neither her muscle nor her liver can metabolise them adequately, so that they accumulate as triacylglycerol, disrupting liver and muscle function.
Carnitine was measured in the liver and muscle biopsy samples. The results were as follows:
CUD |
reference range |
|
| plasma carnitine (µmol /L) | 5.1 |
40 - 60 |
| muscle carnitine (mmol /kg) | 0.002 |
2 - 3 |
| liver carnitine (mmol /kg) | 0.01 |
800 - 1500 |
What conclusions can you draw from these results?
The normal intracellular concentrations of carnitine are very much higher than that in plasma - muscle carnitine is some 50-fold higher than plasma carnitine, and in liver the concentration is more than 10,000-times that in plasma.
We can assume that the very high concentration of carnitine in liver results from both dietary intake (the liver is likely to buffer the concentration that is made available to the rest of the body), and also the fact that carnitine is synthesised in the liver.
The fact that muscle accumulates carnitine to a concentration that is so much higher than that in plasma suggests that there must be active transport of carnitine into muscle.
We already know that CUD is carnitine deficient because of excessive urinary losses, and this may explain the very low concentrations in her muscle and liver.
Cultured fibroblasts and myoblasts accumulate carnitine in a saturable manner. This uptake is inhibited by incubation under anaerobic conditions, the addition of cyanide (a respiratory poison), 2,4-dinitrophenol (an uncoupler of electron transport) and oligomycin (an inhibitor of ATP synthase), or incubation under conditions of sodium ion depletion. (See the exercise on Overheating after overdosing on E - and slimming by taking dinitrophenol to revise the actions of these compounds).
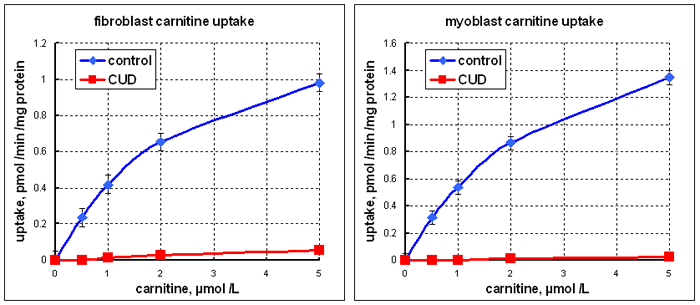
From data reported by Pons et al. Pediatric Research 42: 583 1997
What conclusions can you draw from these observations?
This confirms that muscle and fibroblasts take up carnitine by active transport; the inhibition by sodium ion depletion suggests that the carnitine transporter is sodium-dependent .
The graph on the right above shows uptake of radioactive carnitine into cultured fibroblasts and myoblasts from control subjects and CUD.
What conclusions can you draw from these results?
This suggests that CUD's problem is in the carnitine transporter, not only in the renal tubules, but also in muscle and other tissues. The kidney transporter prevents loss of carnitine in the urine; the transporter in other tissues permits uptake of carnitine to a considerably higher concentration than that in plasma.
The liver does not have the same high affinity carnitine transporter as kidney, muscle and other tissues, but has a low affinity transporter that will permit it to take up or secrete carnitine.
It is highly unlikely that she would have defects in two different carnitine transporters. Why do you think her liver carnitine was so low?
This presumably reflects her very low plasma concentration of carnitine as a result of the continual loss in her urine.
Similar problems of early fatigue, muscle weakness and profound fasting hypoglycaemia with undetectable ketone bodies occur in patients treated for prolonged periods with with the antibiotic pivampicillin.
Pivampicillin is the antibiotic ampicillin (a penicillin-like beta-lactam antibiotic) that has been conjugated with pivalic acid to increase its absorption and bio-availability when given orally. In the liver the conjugate is hydrolysed, releasing active ampicillin and pivalic acid, as shown below.

Like other non-esterified fatty acids, the pivalic acid is esterified with coenzyme A, but cannot be metabolised; there is no pathway for pivalic acid or pivaloyl CoA metabolism in human tissues.
We saw in the exercise on Poisoned by unripe ackee fruit that there is only a small amount of CoA in tissues, and it turns over rapidly. The problem with unripe ackee fruit was that the metabolite of hypoglycine esterified to coenzyme A depleted tissue levels of CoA to such an extent that fatty acid metabolism and oxidative metabolism of pyruvate arising from glycolysis were severely impaired, leading to profound hypoglycaemia.
Here the problem does not seem to be sequestration of CoA, since the pivaloyl moiety can be transferred from CoA onto carnitine, forming pivaloyl carnitine, which is excreted in the urine. However, this can lead to depletion of tissue reserves of carnitine, and eventually to carnitine deficiency.
This provides an easy way of investigating carnitine deficiency, and hence carnitine function, in experimental animals. In the following studies, rats were fed a carnitine-free diet and dosed with pivalic acid to induce carnitine deficiency. After the animals were killed, studies were performed using the gastrocnemius muscle and isolated liver cells.
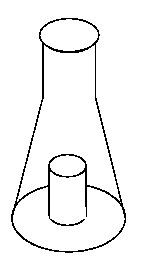 In
the first set of experiments portions of gastrocnemius muscle were incubated
with three different fatty acids, all labelled with [14C] in carbon-1 at a specific
radioactivity of 1 µCi /µmol, with and without the addition of carnitine.
The tissue samples were incubated in centre well vials, and after 20 min, 1
mL of methoxymethylamine was injected through the seal into the outer compartment
of the incubation vial, to trap carbon dioxide, and 0.5
mL of 1 mol /L perchloric acid was injected into the incubation mixture in the
centre well, to precipitate proteins, and drive carbon dioxide out of the solution.
In
the first set of experiments portions of gastrocnemius muscle were incubated
with three different fatty acids, all labelled with [14C] in carbon-1 at a specific
radioactivity of 1 µCi /µmol, with and without the addition of carnitine.
The tissue samples were incubated in centre well vials, and after 20 min, 1
mL of methoxymethylamine was injected through the seal into the outer compartment
of the incubation vial, to trap carbon dioxide, and 0.5
mL of 1 mol /L perchloric acid was injected into the incubation mixture in the
centre well, to precipitate proteins, and drive carbon dioxide out of the solution.
The flasks were shaken for a further 60 min, then the methoxymethylamine (containing carbon dioxide) was washed out with scintillator fluid and transferred to a scintillation counter vial.
During the incubation the muscle was stimulated electrically, in order to increase ATP utilisation, and hence oxidative metabolism. The results were as follows (figures show dpm in carbon dioxide /gram tissue for 3 x replicate incubations):
| fatty acid | no carnitine added |
+ 3 mmol /L carnitine |
| butyrate (C4:0) | 2400 ± 150 |
2390 ± 140 |
| caprate (C10:0 | 2390 ± 145 |
2405 ± 160 |
| palmitate (C16:0) | 110 ± 20 |
2510 ± 167 |