Metabolism
on-line - the virtual tutorial room
copyright © 2008 - 2015 David A Bender
Life-threatening acidosis in an alcoholic - and in a hunger striker given intravenous glucose
PC is a 50 year old man, 174 cm tall and weighing 105 kg. He is an engineer, and works on secondment in one of the strict Islamic states in the Gulf, where alcohol is prohibited.
At the beginning of August he returned to England for his annual leave. According to his family, he behaved as he usually did when on home leave, consuming a great deal of alcohol and refusing meals. He was known to be drinking 2 litres of whiskey, two or three bottles of wine, and a dozen or more cans of lager each day; he refused most meals and his solid food consisted mainly of sweets and biscuits.
On September 1st he was admitted to the Emergency Department of University College Hospital, semi-conscious, and with a rapid respiration rate (40 /min). His blood pressure was 90/60 and his pulse rate was 136 /min. His temperature was normal (37.1 °C). Emergency blood gas analysis revealed severe acidosis: pH 7.02 and base excess -23.
(The base excess is the amount of acid required to return blood pH to the normal range of 7.35 - 7.45 - a negative base excess indicates acidosis)
Why do you think he was breathing rapidly?
The usual response to acidosis is to increase the rate of breathing, so as to expel carbon dioxide, and so shift the equilibrium below to the left, lowering the hydrogen ion concentration in the bloodstream, and so raising plasma pH.
![]()
He was transferred to ITU and given intravenous bicarbonate.
Why do you think was he given intravenous bicarbonate?
Providing bicarbonate will permit the equilibrium above to shift to the left, providing carbon dioxide to be exhaled, and so raising plasma pH.
His pulse rate remained high, and his blood pressure low, so emergency cardiac catheterisation was performed; this revealed a cardiac output of 23 litres /min (normal 4 - 6). A chest X-ray show significant cardiac enlargement.
Why do you think his blood pressure was so low?
Both alcohol and acidosis cause vasodilatation, so that the blood pressure falls as blood is pumped by the heart into a arteries that are wider, and so offer less resistance to blood flow.
Why do you think his pulse rate and cardiac output were so high?
This is to compensate for the lower blood pressure caused by vasodilatation - pumping faster, and with a greater stroke volume, will permit a higher blood pressure to be achieved.
Why do you think he showed cardiac enlargement?
This is again compensation / adaptation. We have to assume that he has been drinking heavily for a considerable time (probably also when not on leave, even though it was illegal). Consequently his heart has been pumping harder and faster in an attempt to maintain blood pressure, and this has led to hypertrophy of cardiac muscle and hence enlargement of his heart.
The following results (all as mmol /L) were reported by the Clinical Chemistry laboratory, on a blood sample taken when he arrived in the Emergency Department:
PC |
reference range |
|
| glucose | 7.6 |
3.5 - 5.0 |
| sodium | 142 |
131 - 151 |
| potassium | 3.9 |
3.4 - 5.2 |
| chloride | 91 |
100 - 110 |
| bicarbonate | 5.0 |
21 - 29 |
| lactate | 18.9 |
0.9 - 2.7 |
| pyruvate | 2.5 |
0.1 - 0.2 |
His fasting blood glucose is above normal, which might indicate diabetes mellitus (which might be expected in an obese middle aged man), but we have no further evidence, and the most striking abnormalities are the low chloride and bicarbonate and very high lactate and pyruvate.
The low plasma bicarbonate is the result of hyperventilation in an attempt to compensate for the acidosis, as discussed above.
Why do you think his plasma chloride was low?
The total concentration of anions must balance the total concentration of cations. Since sodium and potassium are normal, and lactate and pyruvate are abnormally high, plasma chloride is low to maintain electrochemical neutrality.
What we have to think about is why his plasma lactate and pyruvate are so very high. His plasma lactate is approximately 10-fold higher than normal, and his plasma pyruvate approximately 16-fold higher than normal.
(This is based on a true case in UCLH / The Middlesex Hospital, and I am grateful to Dr Hugh Montgomery, who was Consultant in ICU at the time, for drawing it to my attention.)
- and in a hunger striker given intravenous glucose
BD is a political prisoner, and in protest against what she considers to be unlawful imprisonment by a repressive military dictatorship, she has been on hunger strike for the last 6 weeks - she has been refusing all food, but has drunk water (if not, she would not have survived more than a few days). Her case has attracted a considerable amount of international publicity, and she now weighs only 30 kg. There are fears that she is close to death, and her jailers have put her on an intravenous glucose drip in an attempt to save her life.
Shortly after the intravenous glucose began to be administered she began to hyperventilate. Again emergency blood gas analysis revealed severe acidosis: pH 7.12 and base excess -19.
The following results on a blood sample (all as mmol /L) were reported by the Clinical Chemistry laboratory of the prison hospital:
BD |
reference range |
|
| glucose | 9.6 |
* |
| sodium | 141 |
131 - 151 |
| potassium | 4.1 |
3.4 - 5.2 |
| chloride | 90 |
100 - 110 |
| bicarbonate | 5.2 |
21 - 29 |
| lactate | 16.7 |
0.9 - 2.7 |
| pyruvate | 2.3 |
0.1 - 0.2 |
*The reference range for plasma glucose is not relevant in someone who is being given intravenous glucose.
The metabolic acidosis seems to be the same as in PC - very elevated plasma concentrations of lactate and pyruvate, with a low bicarbonate because of respiratory compensation for the acidosis.
It is noteworthy that in both of these patients both lactate and pyruvate are considerably higher than normal - in later exercises you will consider problems associated with elevated plasma lactate, but normal plasma pyruvate.
How is lactate normally metabolised?
In the previous studies of glucose metabolism in muscle, we were working with a high speed supernatant preparation - essentially cytosol. The table below shows the results of incubating a low speed supernatant of muscle homogenate (i.e. one that contains mitochondria but no nuclei or cell debris) with [14C-U]glucose at 1 µCi /mmol, either anaerobically or in the presence of oxygen. As in previous studies, each incubation contained the supernatant from 1 g of muscle.
The incubations contained
200 µmol glucose
20 µmol ATP
20 µmol NAD
600 µmol ADP
200 µmol sodium phosphate
After incubation at 30C for 30 min, incubations were stopped by adding trichloroacetic acid to denature proteins, centrifuged and the supernatant was neutralised with sodium hydroxide. Aliquots of the supernatant were then subjected to high pressure liquid chromatography to measure both the amounts of various metabolites present and and the radioactivity in each. The results are shown as total amount of each metabolite and the specific radioactivity (µCi / mmol) - mean values ± sd from 3 x replicate incubations.
The following results were obtained:
aerobic |
anaerobic |
|||
µmol |
µCi /mmol |
µmol |
µCi /mmol |
|
| glucose | 100 ± 1.5 |
1.01 ± 0.03 |
98 ± 2.5 |
0.99 ± 0.02 |
| pyruvate | 6.2 ± 0.2 |
0.49 ± 0.02 |
6.5 ± 0.2 |
0.51 ± 0.03 |
| lactate | 5.69 ± 0.3 |
0.48 ± 0.03 |
160 ± 5 |
0.48 ± 0.02 |
| carbon dioxide | 80 ± 1.2 |
0.17 ± 0.01 |
0 |
- |
What conclusions can you draw from these results?
Under anaerobic conditions, lactate accumulates, in the same way as it does when a high speed supernatant, not containing mitochondria, is incubated with [14C-U]glucose. However, the experiments with the high speed supernatant were conducted under aerobic conditions, not anaerobic.
Under aerobic conditions little lactate accumulates - it is almost all oxidised to carbon dioxide and water. The specific activity of the label in carbon dioxide is about 1/6 of that in glucose, which is what would be expected. 1 mol of glucose will give rise to 6 mol of carbon dioxide, and in [14C-U]glucose there is label in all 6 carbon atoms, so each carbon atom has 1/6 of the total label in the glucose.
This suggests that lactate undergoes onward oxidation in the mitochondria to yield carbon dioxide.
Further studies, using a manometer, showed that oxidation of 1 mol of lactate to carbon dioxide was associated with consumption of 4.5 mol oxygen.
Is the problem beriberi due to thiamin deficiency?
Beriberi is the classical disease due to deficiency of thiamin (vitamin B1). There are three main forms:
Dry beriberi
Chronic deficiency of thiamin, especially associated with a high carbohydrate diet, results in beriberi, which is a symmetrical ascending peripheral neuritis. Initially the patient complains of weakness, stiffness and cramps in the legs, and is unable to walk for more than a short distance. There may be numbness of the dorsum of the feet and ankles, and vibration sense may be diminished. As the disease progresses, the ankle jerk reflex is lost, and the muscular weakness spreads upwards, involving first the extensor muscles of the foot, then the muscles of the calf, and finally the extensors and flexors of the thigh. At this stage there is pronounced toe and foot drop – the patient is unable to keep either the toe or the whole foot extended off the ground. When the arms are affected there is a similar inability to keep the hand extended – wrist drop.
The affected muscles become tender, numb and hyperaesthetic. The hyperaesthesia extends in the form of a band around the limb, the so-called stocking and glove distribution, and is followed by anaesthesia. There is deep muscle pain, and in the terminal stages, when the patient is bed-ridden, even slight pressure, as from bed clothes, causes considerable pain.
Wet beriberi
The heart may also be affected in beriberi, with dilatation
of arterioles, rapid blood flow and increased pulse rate and pressure, and increased
jugular venous pressure leading to right-sided heart failure and oedema –
so-called 'wet' beriberi.
The signs of chronic heart failure may be seen without peripheral neuritis.
The arteriolar dilatation, and possibly also the oedema, probably results from
high circulating concentrations of lactate and pyruvate.
Acute pernicious (fulminating) beriberi – shoshin beriberi
Heart failure without increased cardiac output, and no peripheral oedema, may also occur acutely, associated with severe lactic acidosis. This was historically a common presentation of thiamin deficiency in Japan, where it was called shoshin (= acute) beriberi; in the 1920s some 26,000 deaths a year were recorded. With improved knowledge of the cause, and improved nutritional status, the disease has become more or less unknown, although it occurs among alcoholics, when the lactic acidosis may be life threatening, without clear signs of heart failure.
Both PC and TD seem to be suffering from acute pernicious beriberi.
Early studies of patients with beriberi showed high plasma concentrations of lactate and pyruvate, which fell to normal on treatment with thiamin.
In order to understand how thiamin deficiency leads to elevated plasma lactate and pyruvate, and therefore how thiamin is normally involved in glucose metabolism we can look at some of the classical studies conducted by Peters and colleagues in the 1920s and 1930s.
 (If
you want to read more about these early studies, they were summarised in: Peters
RA, Biochemical lesions and lethal synthesis, Pergamon Press, Oxford, 1963).
(If
you want to read more about these early studies, they were summarised in: Peters
RA, Biochemical lesions and lethal synthesis, Pergamon Press, Oxford, 1963).
Peters used pigeons in his studies of thiamin deficiency - they show a characteristic head retraction when they are fed on a high carbohydrate diet with no thiamin, and restoration of the vitamin leads to rapid normalisation.
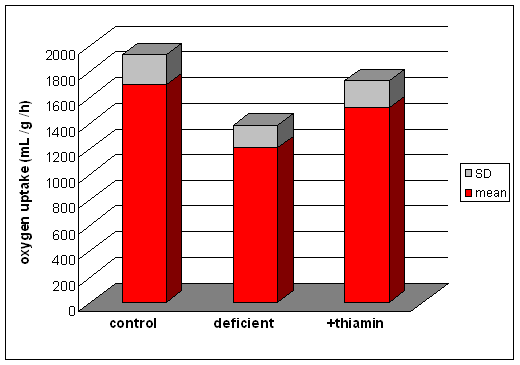 They measured
oxygen uptake by brain homogenates from control pigeons, those maintained on
a thiamin deficient high carbohydrate diet (the birds showed head retraction)
and deficient animals that had been repleted with thiamin (in which the head
retraction was cured).
They measured
oxygen uptake by brain homogenates from control pigeons, those maintained on
a thiamin deficient high carbohydrate diet (the birds showed head retraction)
and deficient animals that had been repleted with thiamin (in which the head
retraction was cured).
When the brain homogenates were incubated with 0.37 mmol /L lactate, the results were as shown on the right.
(From data reported by Gavrilescu N et al, Proceedings of the Royal Society B 10: 431 (1932)
When they incubated brain homogenates with succinate or malate, two substrates that are known to undergo complete oxidation to carbon dioxide and water, there was no difference between the control and deficient animals.
What conclusions can you draw from these results?
Succinate and malate are two intermediates of the citric aid cycle that undergo complete oxidation to carbon dioxide and water. since their oxidation is unaffected by thiamin deficiency, it is reasonable to assume that citric acid cycle activity is unimpaired. Therefore the problem must lie in the oxidation of pyruvate.
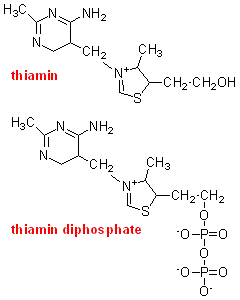 Although
the administration of thiamin to deficient animals led to a rapid recovery,
studies with brain homogenates showed that the addition of thiamin in vitro did not increase pyruvate oxidation and oxygen uptake. In brains from deficient
animals that had been repleted with [32S]thiamin there was accumulation of radioactivity,
but mainly as thiamin diphosphate.
Although
the administration of thiamin to deficient animals led to a rapid recovery,
studies with brain homogenates showed that the addition of thiamin in vitro did not increase pyruvate oxidation and oxygen uptake. In brains from deficient
animals that had been repleted with [32S]thiamin there was accumulation of radioactivity,
but mainly as thiamin diphosphate.
What conclusions can you draw from this observation?
How might you test your hypothesis?
It seems likely that it is thiamin diphosphate rather than thiamin itself that is the coenzyme required for the oxidation of pyruvate. This could be tested by incubating a mitochondrial preparation from thiamin deficient brain with thiamin disphosphate and measuring oxygen consumption. The results of such an experiment are show below.
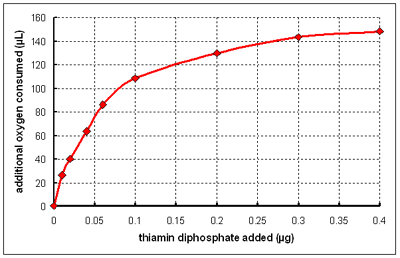
(From data reported by Banga I et al, Biochemical Journal 33: 1109-21 1939)
If isolated mitochondria are incubated with [14C-U] pyruvate then all of the radioactive label appears in carbon dioxide. If the mitochondria have been pre-treated with cyanide, which inhibits oxygen consumption, there is little or no release of radioactive carbon dioxide.
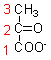 However,
in the presence of added NAD, cyanide-treated mitochondria do metabolise pyruvate,
releasing radioactive carbon dioxide from [14C-1]pyruvate, but not from [14C-2]
or [14C-3]pyruvate - and the amount of carbon dioxide released is proportional
to the amount of NAD added.
However,
in the presence of added NAD, cyanide-treated mitochondria do metabolise pyruvate,
releasing radioactive carbon dioxide from [14C-1]pyruvate, but not from [14C-2]
or [14C-3]pyruvate - and the amount of carbon dioxide released is proportional
to the amount of NAD added.
What conclusion can you draw from these results?
It seems likely that pyruvate undergoes both a decarboxylation, losing carbon -1 as carbon dioxide and also an oxidation. The likely product of such a reaction would be acetate. In fact, free acetate is not formed; the product is acetyl CoA - acetate esterified to coenzyme A.
There is a further intermediate step in the reaction that you could not deduce from the experiments reported so far. The immediate acceptor of the hydrogen released in the oxidation of pyruvate to acetate is not NAD but the disulphide compound lipoamide. Reduced lipoamide is reoxidised in a later part of the reaction sequence, by reduction of NAD to NADH.
The full sequence of the reaction, which is catalysed by a large multi-enzyme complex, is shown below.
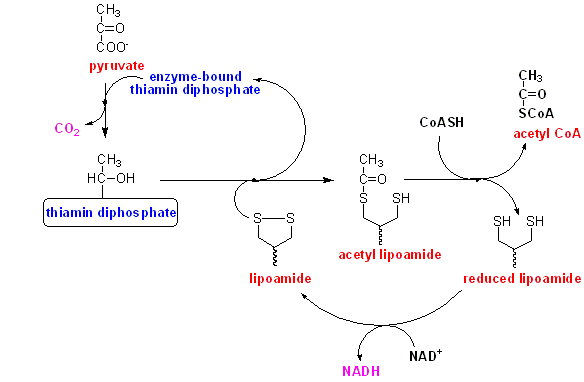
Pyruvate reacts with enzyme-bound thiamin diphosphate, undergoing decarboxylation to hydroxy-ethyl thiamin diphosphate.
The hydroxy-ethyl group is transferred to enzyme-bound lipoamide, forming acetyl lipoamide.
The acetyl group s transferred onto coenzyme A, forming acetyl CoA, and leaving reduced lipoamide at the active site of the enzyme.
Reduced lipoamide is reoxidised by reaction with NAD.