Metabolism
on-line - the virtual tutorial room
copyright © 2008 - 2015 David A Bender
Fasting hypoglycaemia in an infant - and poor exercise tolerance in two brothers
LR is a 16 month old baby, who was born at term after an uneventful pregnancy. Until she was 15 months old she had been fed every 3 - 4 hours, and never slept though the night, but woke every 3 - 4 hours, demanding to be fed.
At 15 months of age she slept through the night for the first time, and was found at 6 am in a generalised tonic-clonic seizure, which might suggest epilepsy. On admission to hospital a blood sample was taken; her plasma glucose was 1.5 mmol /L (the reference range for a fasting infant is 3.9 - 6.1 mmol /L), and a urine dipstick test showed the presence of a high concentration of ketone bodies.
What is the most likely cause of her seizure?
Almost certainly the very low plasma concentration of glucose. After a normal overnight fast her plasma glucose is as low as might be expected after several days of starvation, and you would not expect a very high level of ketone bodies in the urine after an overnight fast.
One possible cause of her abnormally low fasting plasma glucose would be an excessive and inappropriate secretion of insulin, but when her plasma insulin was measured it was at an appropriate level for her plasma glucose concentration in both the fed and fasting states.
She suffered a further seizure a few weeks later, again when she had slept through the night, and had not woken demanding to be fed.
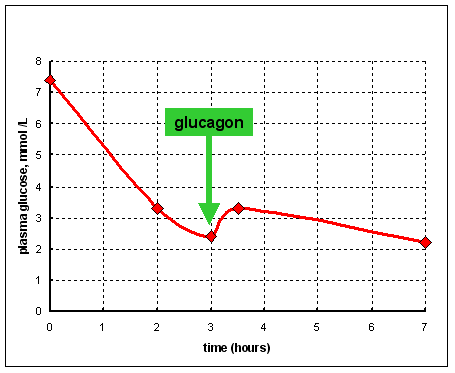 Her blood
glucose and lactate were measured at intervals for 7 hours after feeding, and
for a further 2.5 hours after breakfast. After 3 hours of fasting she was given
0.36 mg of glucagon intravenously.
Her blood
glucose and lactate were measured at intervals for 7 hours after feeding, and
for a further 2.5 hours after breakfast. After 3 hours of fasting she was given
0.36 mg of glucagon intravenously.
The results in the fasting state are shown on the right (from data reported by Rutledge SL et al. Pediatrics 108 495-7 2001).
What effect would you expect an injection of glucagon to have on plasma glucose in the fasting state?
Glucagon has two key actions:
stimulation of glycogen breakdown in the liver to release glucose
stimulation of gluconeogenesis from amino acids
Therefore you would expect to see an increase in plasma glucose. However, LR shows only a minute increase in plasma glucose after an injection of glucagon that would have been expected to lead to a considerable increase.
On another occasion she was given an injection of adrenaline instead of glucagon. Again there was no significant increase in her plasma concentration of glucose.
What conclusions can you draw from these results?
Adrenaline would also be expected to raise blood glucose, again by stimulating breakdown of liver glycogen.
This suggests several possibilities:
She may be unable to mobilise liver glycogen because of defects in her adrenaline and glucagon receptors. This is unlikely, since it is unlikely that two different hormone receptors would both be defective.
She may be unable to mobilise liver glycogen because of a defect in the downstream signalling from her adrenaline and glucagon receptors - both hormones act via a cyclic AMP second messenger system, so a defect in responses to cyclic AMP is a possibility.
How might you test this?
You could measure plasma non-esterified fatty acids. The concentration will increase in response to adrenaline, which stimulates hormone-sensitive lipase in adipose tissue. When this was done, her response to adrenaline was normal, suggesting that the problem is not in the downstream signalling in response to the hormones.
The remaining possibilities are that:
She is unable to synthesise glycogen because of lack of one or other of the enzymes involved in glycogen synthesis.
She is unable to break down glycogen and release glucose because of lack of one or other of the enzymes involved in glycogen breakdown.
What would you expect to observe in someone who could synthesise glycogen but not break it down?
In someone who can synthesise glycogen but not break it down, you would expect to see accumulation of glycogen in the liver, to such an extent that her liver would be enlarged and readily palpable. There is no indication that her liver is enlarged.
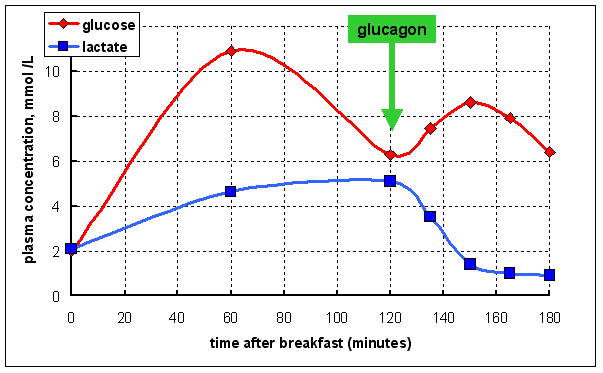 In
a further study her plasma glucose and lactate were measured after a breakfast
of milk and cereal with sugar; 2 hours after breakfast she was again given an
intravenous injection of 0.36 mg of glucagon
In
a further study her plasma glucose and lactate were measured after a breakfast
of milk and cereal with sugar; 2 hours after breakfast she was again given an
intravenous injection of 0.36 mg of glucagon
You would not expect plasma glucose to rise above about 8 mmol /L in an infant after a normal small breakfast.
What conclusions can you draw from these results?
Normally, in response to insulin after a meal you would expect much of the glucose entering from the gut to be taken up by the liver and used for glycogen synthesis, so that there is only a modest rise in plasma glucose. The very high plasma glucose suggests that this is not happening.
More significantly, the increase in plasma lactate after a meal suggests that glucose is being metabolised, but largely by anaerobic glycolysis.
The response to glucagon is an increase in plasma glucose, but largely at the expense of lactate, suggesting that she is able to convert lactate to glucose, but cannot synthesise glycogen from that glucose.
In a further study she was given an oral dose of 1 g of galactose /kg body weight after an overnight fast. Galactose is phosphorylated to galactose 6-phosphate by hexokinase, and can then be isomerised to glucose 6-phosphate. This may be metabolised or may be used to synthesise glycogen.
 Plasma
glucose, lactate and ketone bodies were measured, as shown in the graph on the
right. (From data reported by Aynsley-Green A et al. Arch Dis Childhood 52:
573-9 1977)
Plasma
glucose, lactate and ketone bodies were measured, as shown in the graph on the
right. (From data reported by Aynsley-Green A et al. Arch Dis Childhood 52:
573-9 1977)
What conclusions can you draw from these results?
She is obviously able to metabolise galactose, and use it as a source of glucose. The fall in plasma ketone bodies is presumably the result of inhibition of ketogenesis as a result of the increase in plasma glucose.
Again she seems to be able to metabolise galactose, but with production of much lactate. As previously this suggests that she is unable to synthesise glycogen.
At this stage in the investigation it would probably be appropriate to perform liver and muscle biopsies to measure glycogen, and glycogen synthase, the key enzyme of glycogen synthesis.
Why do you think this should be done a couple of hours after a meal?
Glycogen is synthesised in the fed state and utilised in the fasting state, so in the fasting state there might be very little glycogen present anyway.
The results obtained were as follows:
glycogen (g /100 g tissue) |
glycogen synthase (µmol
glucose incorporated /min /g tissue) |
|||
LR |
control subjects |
LR |
control subjects |
|
| liver | 0.65 |
2.0 - 6.0 |
0.04 |
1.99 - 3.60 |
| muscle | 0.76 |
0.72 - 0.75 |
1.71 |
1.51 - 1.85 |
(From data reported by Aynsley-Green A et al. Arch Dis Childhood 52: 573-9 1977)
What conclusions can you draw from these results?
Her problem is obviously very low activity of glycogen synthase in her liver, so that in the fed state she cannot build up reserves of glycogen to maintain blood glucose in the fasting state. This means that after an overnight fast she is profoundly hypoglycaemic and has an abnormally high plasma concentration of ketone bodies.
Interestingly, her muscle glycogen synthase is within the normal range, and she has a normal level of muscle glycogen.
What conclusion can you draw from this?
This suggests that liver and muscle glycogen synthase are different enzymes, coded for by different genes.
How might you investigate whether this is a genetic defect in her liver glycogen synthase?
One way would be to look for any other affected individuals in her family and see whether there is a clear pattern of inheritance. In this case there are no affected relatives.
The alternative approach would be to take liver biopsy samples from her parents and measure glycogen synthase. Since they are unaffected, if the condition is genetic then LR must be homozygous for defective glycogen synthase, and they must be heterozygous, and would be expected to have about half the normal activity of the enzyme in their livers.
The pathway of glycogen synthesis is shown below. Note that UTP is equivalent to ATP, so that for each mol of glucose added to glycogen there is a cost equivalent to 2 x ATP.

What do you think is the significance of the reaction of pyrophosphatase, which hydrolyses the pyrophosphate released by uridyl transferase to 2 mol of inorganic phosphate?
This is a fairly common occurrence in metabolism - pyrophosphatase removes pyrophosphate, one of the products of the uridyl transferase reaction, so that it not available to undergo the reverse reaction. This means that the reaction of uridyl transferase is irreversible under normal conditions.
The importance of this is that, as you will see in later exercises, glycogen synthesis and utilisation are closely regulated, and as we have seen in the exercise on Breathless after sprinting, it is common to have irreversible steps in pathways that are regulated reciprocally.
Lack of glycogen synthase is one of a number of disorders of glycogen metabolism that are collectively known as glycogen storage diseases. This is classified as glycogen storage disease type 0a (affecting the liver).
- and poor exercise tolerance in two brothers
MA and BA are brothers; their parents are first cousins. All their lives they have been unable to keep up with their friends in sport and other physical activity, because they have tired very easily. When MA was 10½ years old he collapsed in the school playground as a result of cardiac arrest, and could not be resuscitated. At post-mortem his heart was found to weigh 200 g (the normal range for a boy of his age is 139 - 178), and his left ventricular wall was 1.7 cm thick (the normal value is < 0.9 cm). The cause of death was listed as hypertrophic cardiomyopathy.
Two years after his brother's death, when he was 11 years old, BA was investigated. Electrocardiography and echocardiography showed flattening of the T-wave and mild generalised hypertrophy of the left ventricle, as well as left atrial enlargement and mildly impaired diastolic function at rest.
He was subjected to exercise tests on a treadmill. In the first the exercise started at a work load of 30 watts, both the speed of the treadmill and the angle of inclination were increased, so as to increase the work required by 10 watts per minute. He was able to continue this for 3.5 minutes, but then gave up due to leg fatigue.
BA |
normal value or range |
|
| workload (W /kg body weight) | 1.8 |
> 3.0 |
| peak heart rate (beats /min) | 200 |
> 190 |
| systolic blood pressure (mm Hg) | increase from 105 to 115 |
increase > 25 |
| peak plasma lactate (mmol /L) | 3.1 |
> 5.0 |
(From data reported by Kollberg G et al. New England Journal of Medicine 357: 1507-14 2007)
What conclusions can you draw from these results?
Where has the lactate come from in this vigorous exercise?
BA has poor exercise tolerance and a low workload; his peak heart rate during exercise is within the normal range, but he is unable to increase his blood pressure as much as normal, suggesting impaired cardiac function.
As we saw in the exercise on Breathless after sprinting, in maximum exertion muscle metabolises relatively anaerobically, putting out lactate which is used in the liver for gluconeogenesis. Some of the glucose that is metabolised in muscle comes from blood glucose, but much comes fm muscle glycogen.
In the second exercise test the work required was increase more gradually, at a rate of 5 watts every 3 minutes . He was able to continue this for 10.5 minutes, but then gave up due to leg fatigue and a drop in blood pressure.
BA |
normal value or range |
|
| workload (W /kg body weight) | 1.2 |
> 3.0 |
| heart rate response (beats /min) | 180 at 3 min of 0.8 W /kg |
< 150 |
6 min plateau at 155 |
steady increase |
|
166 at peak exercise |
> 190 |
|
| systolic blood pressure (mm Hg) | decrease from 115 to 80 |
increase > 25 |
| peak respiratory quotient (CO2 : O2) | 0.86 |
1.0 |
| maximum oxygen uptake (mL /kg body weight /min) | 30 |
> 45 |
| exercise echocardiography, stroke volume | 30% decrease |
increase > 10% |
(From data reported by Kollberg G et al. New England Journal of Medicine 357: 1507-14 2007)